Technology focus: MERFISH#
This notebook will present a rough overview of the plotting functionalities that spatialdata implements for MERFISH data.
Loading the data#
Please download the data from here: MERFISH dataset and adjust the variable containing the location of the .zarr file.
merfish_zarr_path = "./merfish.zarr"
import spatialdata as sd
merfish_sdata = sd.read_zarr(merfish_zarr_path)
merfish_sdata
SpatialData object with:
├── Images
│ └── 'rasterized': SpatialImage[cyx] (1, 522, 575)
├── Points
│ └── 'single_molecule': DataFrame with shape: (3714642, 3) (2D points)
├── Shapes
│ ├── 'anatomical': GeoDataFrame shape: (6, 1) (2D shapes)
│ └── 'cells': GeoDataFrame shape: (2399, 2) (2D shapes)
└── Table
└── AnnData object with n_obs × n_vars = 2399 × 268
obs: 'cell_id', 'region'
uns: 'spatialdata_attrs': AnnData (2399, 268)
with coordinate systems:
▸ 'global', with elements:
rasterized (Images), single_molecule (Points), anatomical (Shapes), cells (Shapes)
Visualise the data#
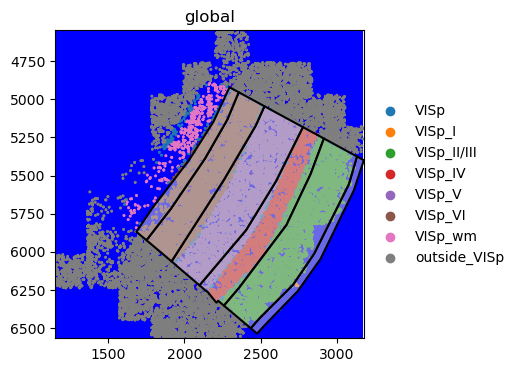
We’re going to create a naiive visualisation of the data, overlaying the annotated anatomical regions contained in anatomical and the tissue image. For this, we need to load the spatialdata_plot library which extends the sd.SpatialData object with the .pl module. Furthermore, we will only select the elements we want to plot using pp.get_elements().
import spatialdata_plot
merfish_sdata.pp.get_elements(["anatomical", "rasterized"]).pl.render_images().pl.render_shapes(
fill_alpha=0.5, outline=True
).pl.show()
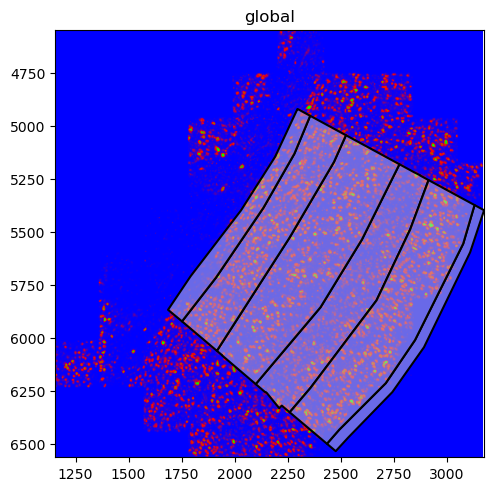
The MERFISH data also contains points which we have so far not visualised. This can be done with the pl.render_points() function. However, since we have over 3 million points, we will only render 1 % of them as to not overplot the image.
merfish_sdata.points["single_molecule"] = merfish_sdata.points["single_molecule"].sample(frac=0.01)
merfish_sdata.pl.render_points().pl.show()
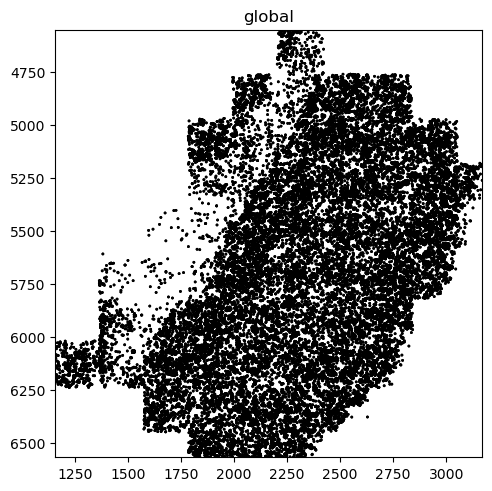
Furthermore, we can overlay all 3 layers and color the points by an annotation.
import matplotlib.pyplot as plt
fig, ax = plt.subplots(ncols=1, figsize=(4, 4))
(
merfish_sdata.pp.get_elements(["anatomical", "rasterized", "single_molecule"])
.pl.render_images()
.pl.render_points(color="cell_type")
.pl.render_shapes(fill_alpha=0.5, outline=True)
.pl.show(ax=ax)
)